
Co-chair, Full-time Instructor (Tenure-Track)
Chemistry Department
Cincinnati State
I believe that all problems and observations in chemistry can be traced back to a simple chemical principle, and I seek to identify those in my research
My research in graduate school was focused on developing and applying computational tools to study reaction mechanisms and chemical trends in biocatalysis. Biocatalysis offers a greener approach to selective transformations over traditional chemical syntheses. Using computational tools, I explored the mechanisms of enzymes and the subsequent transformations and reactivity of their products, so that we can exploit them to aide in expansion of substrate scopes and synthesis of bioactive compounds.
Using tools developed by the Zimmerman Group, I identified the underpinning chemical principle behind the reactivity of benzyl alcohol products produced by the enzymes CitB and ClaD. My analyses not only correlated with experimental outcomes, but where then used to predict more stable analogs that could be isolated for further transformations.

Doyon, T. J.; Perkins, J. C.; Baker Dockrey, S. A.; Romero, E. O.; Skinner, K. C.; Zimmerman, P. M.; Narayan, A. R. H. Chemoenzymatic o-Quinone Methide Formation. J. Am. Chem. Soc. 2019, 141, 20269–20277. DOI: 10.1021/jacs.9b10474
Using small QM models, I explored the mechanism of the enzyme TropC, which catalyzes an unprecedented ring expansion to form an aromatic compound, known as stipitaldehyde. Previously proposed to undergo a polar ring expansion, we instead found that a radical pathway is more suitable for the transformation, and correlates well with experiments.
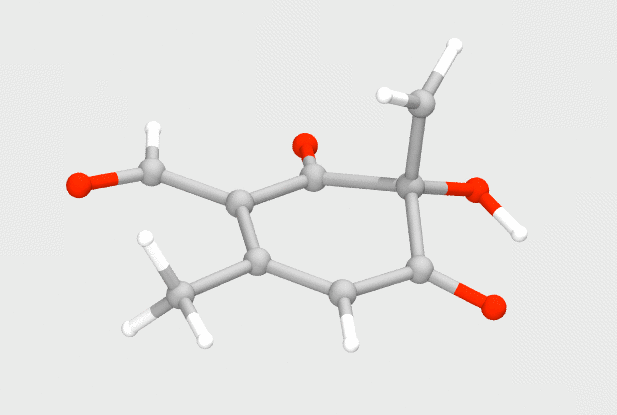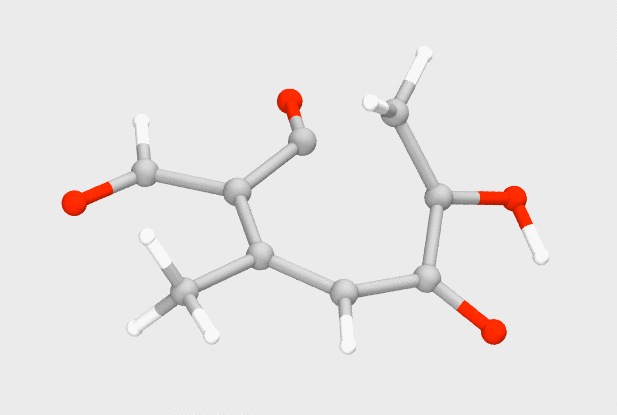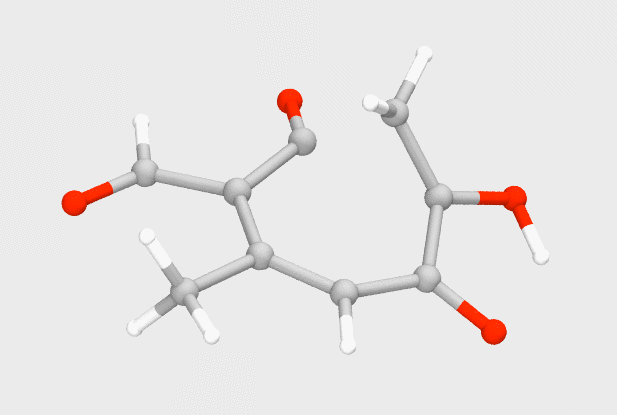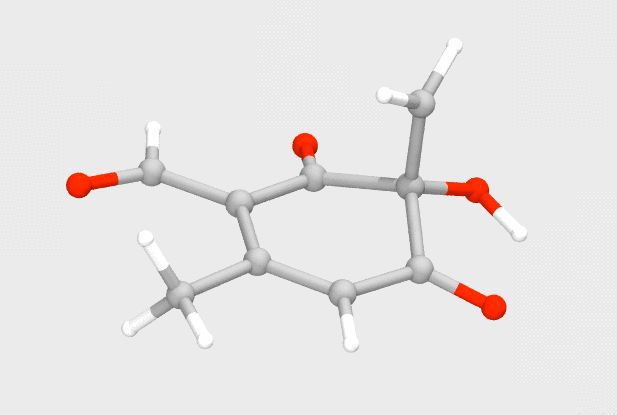
Doyon, T. J.; Skinner, K. C.; Yang, D.; Mallik, L.; Wymore, T.; Koutmos, M.; Zimmerman, P. M.; Narayan, A. R. H. Radical tropolone biosynthesis. ChemRxiv 2020. DOI: 10.26434/chemrxiv.12780044
To successfully study reaction pathways within an enzyme model for TropC, I needed to study both one- and two-electron pathways within a sully solvated TropC enzyme. This resulted in the need for a new computational tool to study single-electron transfers (SETs) along reaction pathways within enzyme active sites. I was able to develop a combined constrained density functional theory/molecular mechanics (CDFT/MM) tool and successfully applied to two simple organic electron donor systems: tetrakis(dimethylamino)ethylene (TDAE) and tetrathiafulvalene (TTF). I was able to replicate known chemistry (rates of rates/energy barriers, spin densities, etc.), refine a mechanistic pathway, and provide insight into chemical trends between structure and thermodynamics and kinetics.

Skinner, K. C.; Kammeraad, J. A.; Wymore, T.; Narayan, A. R. H.; Zimmerman, P. M. Simulating Electron Transfer Reactions in Solution: Radical-Polar Crossover. J. Phys. Chem. B 2023, 127, 10097–10107. DOI: 10.1021/acs.jpcb.3c06120
With the creation of my new CDFT/MM tool, I explored various pathways within a fully solvated TropC enzyme model. Included in these pathways was a SET pathway, which I observed ring expansion to a tautomer of our stipitaldehyde natural product. I also explored other pathways, including the aforementioned radical pathway which was found to also be plausible within the enzyme active site.

Skinner, K. C. Development and Application of Computational Tools to Study Single-Electron Transfer Initiated Reactions in Solution and Enzymes. Ph.D. Dissertation, University of Michigan, Ann Arbor, MI, 2022.
This research has been applied to another system: Wititsuwannakul, T.; Skinner, K. C.; Kammeraad, J. A.; Yang, D.; Narayan, A. R. H.; Zimmerman, P. M. Uncovering the Origins of Selectivity in Non-Heme Iron Dioxygenase-Catalyzed Tropolone Biosynthesis. J. Phsyc. Chem. B 2025, 129, 7766–7783.